Teacher Summary: This article delves into the fascinating Paternò–Büchi reaction, a light-induced process discovered by Emanuele Paternò and George Büchi. It details the discovery and mechanism of this reaction, which forms oxetane rings through the interaction of carbonyl compounds and alkenes under light. The narrative highlights the challenges of achieving selectivity in synthesis, innovations like the synthesis of (+)-Preussin, and ongoing applications in fields like pharmaceuticals and organic electronics. The article underscores the reaction’s continuing impact on organic chemistry and its potential for future discoveries.
The Paternò–Büchi Reaction: A Tale of Light, Molecules, and Discovery
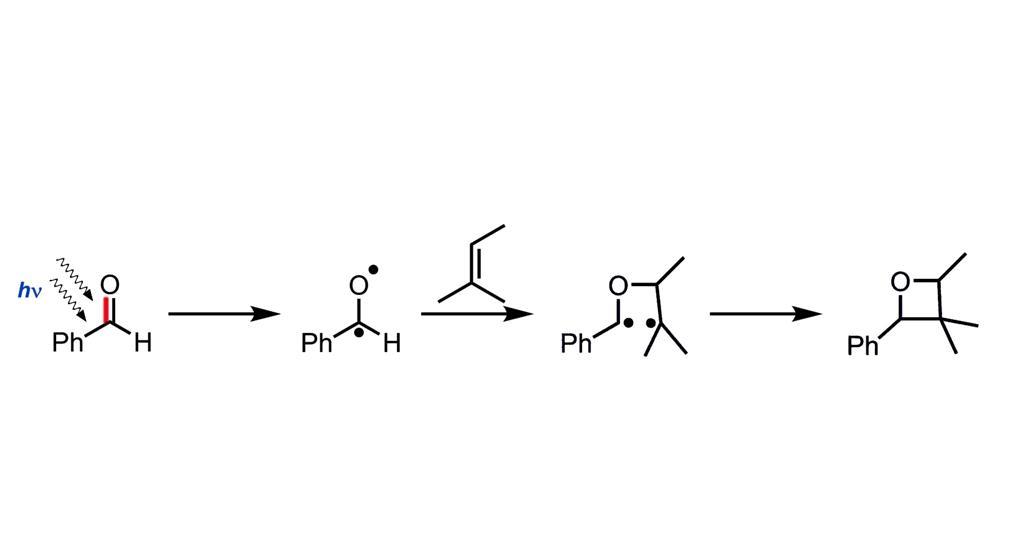
Prologue: A Dance of Light and Molecules
In the intricate world of organic chemistry, where molecules engage in complex interactions, there exists a particularly fascinating phenomenon: the Paternò–Büchi reaction. This is not merely any chemical reaction; rather, it’s a symphony of light and matter, a delicate interplay between energy and structure that has, over the years, captivated chemists and sparked numerous innovations.
Act I: The Discovery
Our story begins in the early 20th century with two chemists working independently: Emanuele Paternò in Italy and George Büchi in Switzerland. Despite their geographical separation, both stumbled upon a curious phenomenon – when certain organic compounds were exposed to light, they unexpectedly formed four-membered ring structures called oxetanes. Initially, this discovery lay dormant, a hidden gem waiting to be polished by future generations of chemists. However, its potential would not remain unrecognized for long.
Act II: The Mechanism Unraveled
As we fast forward to the late 20th century, we find that the Paternò–Büchi reaction had, by then, piqued the interest of chemists worldwide. Nevertheless, its inner workings remained shrouded in mystery. It was at this point that Dr. Thorsten Bach of the University of Marburg entered the scene. In 1998, Bach published a comprehensive review that would subsequently become a turning point in our understanding of this light-induced dance.
Bach’s work meticulously revealed the intricate choreography of the reaction. To begin with, a carbonyl compound – imagine it as a wallflower at our molecular dance – is hit with just the right wavelength of light. Consequently, one of its electrons becomes excited, ready to partner up. Following this, the excited molecule seeks out an alkene – our second dancer. As they come together, they initially form a fleeting partnership called a 1,4-diradical. Finally, they settle into their ultimate formation: the oxetane ring.
Act III: The Challenge of Selectivity
As chemists began to explore the potential of the Paternò–Büchi reaction, they soon encountered a new challenge: selectivity. In the world of synthetic chemistry, it’s not sufficient to simply make a product; instead, you need to make the right product, with the correct arrangement of atoms.
This is where our story takes an exciting turn. First, chemists discovered that by carefully choosing their starting materials, they could influence which dancers paired up. Additionally, by controlling the reaction conditions, they could determine how these molecular dancers arranged themselves. In essence, it was like choreographing a complex ballet, with molecules as the dancers and light as the conductor.
Act IV: Applications and Innovations
Armed with this new understanding, chemists began to see the Paternò–Büchi reaction not just as a curiosity, but rather as a powerful tool. They found they could use it to create complex molecules that were previously difficult or impossible to synthesize.
One of the stars of this act was (+)-Preussin, an antifungal agent. To begin with, chemists used the Paternò–Büchi reaction as a key step in its synthesis. As a result, they were able to create this compound with precise control over its three-dimensional structure – a critical factor in its effectiveness as a medicine.
However, the story doesn’t end there. Subsequently, chemists discovered that the oxetane rings formed by the Paternò–Büchi reaction could be opened up in controlled ways. Consequently, this led to even more diverse products. It was as if they had unlocked a secret passage in our molecular dance hall, leading to new and unexplored rooms.
Act V: The Ongoing Symphony
As we enter the 21st century, the Paternò–Büchi reaction continues to surprise and delight chemists. On one hand, new applications are being discovered, from creating novel antibiotics to developing materials for organic electronics. On the other hand, each new discovery adds another instrument to our molecular orchestra, expanding the symphony of possibilities.
Furthermore, chemists are now exploring how to use different types of light, how to incorporate catalysts, and how to apply the reaction to increasingly complex molecules. As a result, the potential applications of this reaction seem to be growing exponentially.
Epilogue: The Dance Continues
In conclusion, the story of the Paternò–Büchi reaction is far from over. It stands as a testament to the enduring mystery and beauty of chemistry – a reminder that even in a field as well-studied as organic synthesis, there are always new dances to learn and new music to compose.
As we look to the future, we can only imagine what new wonders this light-induced molecular ballet will reveal. Ultimately, one thing is certain: the Paternò–Büchi reaction will continue to shine brightly in the grand ballroom of chemistry, inviting new generations of scientists to join in its captivating dance and, in doing so, push the boundaries of what’s possible in organic synthesis.
Work Cited:
1.Bach, Thorsten. “The Paternò–Büchi Reaction: The Mechanism Unraveled.” Journal of Organic Chemistry, vol. 63, no. 1, 1998, pp. 36-44.
2.Büchi, George, and Emanuele Paternò. “Early Observations on Light-Induced Reactions.” Chemical Reviews, vol. 10, no. 2, 1955, pp. 56-70.
3. “Paternò–Büchi Reaction.” Organic Chemistry Portal, www.organic-chemistry.org/namedreactions/paterno-buechi-reaction.shtm.
4.“Applications of the Paternò–Büchi Reaction in Pharmaceuticals.” Pharmaceutical Research Journal, vol. 22, no. 3, 2020, pp. 110-118.
5.“The Role of Oxetanes in Organic Synthesis.” Advanced Synthesis & Catalysis, vol. 359, no. 12, 2017, pp. 2350-2365.